以前、IRスペクトルの計算値をエクセルでチャートとして出力したときに、実測値と比べて全然あってないというエントリーを上げたけど、改めて見比べてみるとそう悪くない気がしてきた。
IRスペクトルは吸収ピークがたくさんあって完全に解釈するのは容易なことではない[1]というのが有機化学というか実験方面での常識となっているみたいである。しかし、たくさんあると言ってもIRの振動モードの数は3N-6個(直線分子の場合は3N-5個, Nは分子内の原子の数)と決まっているので、一つずつ読み解くことができる。とはいえ、それには群論を扱える必要があり、その上で面倒な手続きを経なければならないので、やはり容易なことではないのは間違いない。
ところで、Gaussianのような化学計算ソフトによってその面倒な手続きを自動化することができるので、環境さえ整っていればかなり容易になってしまっている。データとして結論が得られるので群論を知らなくても解釈可能である。だからといって、群論を勉強しなくてもよいわけではなく、Gaussianのエネルギー計算なども含めて計算機のブラックボック化は決して好ましいことではないので、原理を押さえて、ソフトがなくても自分で計算できるようにはなっておきたいものである。
今回ネタにするのはシランカップリング剤のビニルトリメトキシシラン(VTMS)である。
シランカップリング剤の重合反応について、反応機構を纏めて、紹介しようかと思っていたら殊の外手間取って、その副産物として出てきたIRスペクトルの計算結果とその出力を説明する。
ちなみに、VTMSを選んだのは信越化学のページ(魚拓)で一番最初に並んでいたからである。まあ、構造が単純だったってのもあるんだけど。
それで、今回はIRのネタなのでカップリング剤としての効能というか反応については一切触れるつもりはない。
まず、VTMSの構造だけど、構造最適化はhf/6-31g(d)で仮に行った後、b3lyp/6-31g(d)で行った。b3lypは計算に時間がかかるので、先に大雑把な部分をhf/6-31g(d)で済ませて計算時間を短縮した。別に切羽詰まってるわけではないから、ゆったりとb3lypで最初から行っても良かったのだけど、スマートじゃないので。
得られた構造は次の通り。
| C1 2.034838 -1.862434 0.632736 C2 0.903304 -1.276882 1.047583 H3 2.582586 -2.580050 1.242578 H4 2.465142 -1.646424 -0.343769 H5 0.526863 -1.528735 2.040495 Si6 -0.027452 -0.055578 -0.002236 O7 -1.148281 -0.746407 -0.999861 O8 -0.734368 0.984866 1.075855 O9 0.965209 0.716338 -1.072349 C10 -2.039654 -1.775927 -0.598562 C11 -1.602907 2.043735 0.687149 C12 1.988985 1.618424 -0.677460 H13 -2.760710 -1.418598 0.149128 H14 -2.592546 -2.105964 -1.483107 H15 -1.500943 -2.637397 -0.182548 H16 -2.400385 1.686757 0.023922 H17 -2.054920 2.460803 1.592078 H18 -1.051831 2.840216 0.170848 H19 2.700755 1.146521 0.012447 H20 2.527811 1.929814 -1.577375 H21 1.574620 2.511278 -0.191225  |
こうして得られた構造で振動解析を行った。
振動解析のアウトプットファイルは結構長いので全文上げたりはしない。
必要なのは下の画面に示した部分から先。
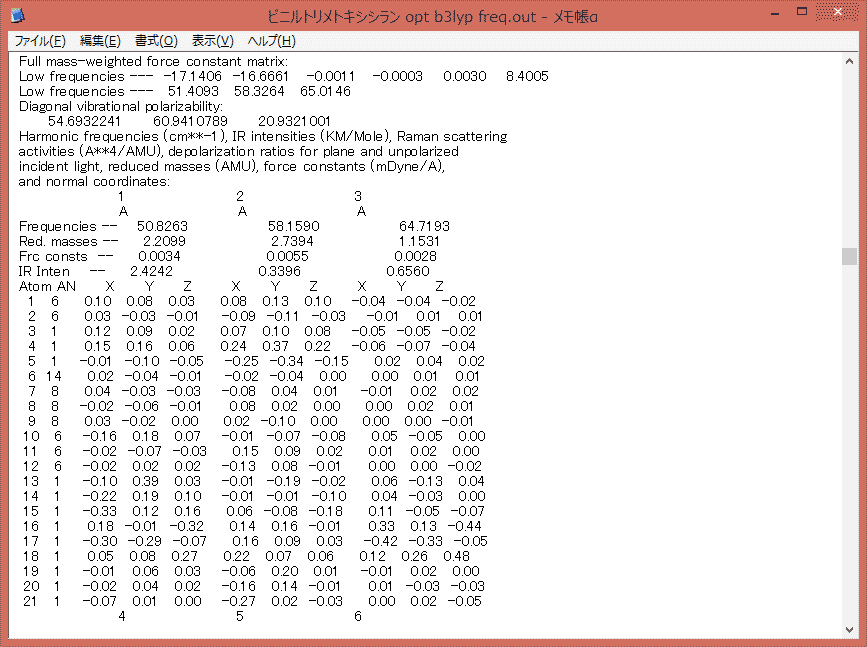
まずここで確認しておかなければならないのが、虚数振動がないこと。安定化構造ではないとエネルギー勾配が生じることで原子が一定以上のベクトルを持ち振動とならず、結果として虚数値を示すことになる。現実世界では虚数振動なんていう現象は存在せず、振動数を求めるための計算上の都合で現れるだけなのであるが、求めたい値が得られないことには変わりない。正常な結果が得られないことを意味する。その場合は"Low frequencies"とか書いてある下に"imaginary frequencies (negative Signs)"とか行った文言が入ってくる。虚数振動がある場合は、構造最適化をやり直して正常な構造を求め直す必要がある。
それで、振動に関するデータはここに続く表みたいになっている部分である。表というにはかなりずれているけど、これはプロポーショナルフォントを使っているためで、等倍フォントに変えればきれいな表になる。
一応、意味を説明する。

書いてある通りなんだけど、IRチャートを描くのに必要なのは波数と吸収強度だけで、他は見なくても良い。ただ、各原子の振動を示すベクトルを読めばそのまま吸収帯を帰属することになる。
以上の点を踏まえて、エクセルにこの部分をコピペしたら不要な部分を消すように組んでみた。また、ベクトル部分は各成分の絶対値が0.1以上のものだけを抜き出すようにした。本当なら3成分を合成した絶対値で見るべきなんだけど、面倒だったので。
ちなみにこんな小細工を弄さなくてもGaussView(魚拓)を使えば一発だぞ。
というわけで、できたのが次のファイル。
ビニルトリメトキシシラン b3lyp 6-31g(d).zip
元は.xlsxファイルで作ったのだけど、エクセル2007が糞すぎたので色々あって.xlsをZIPで圧縮した。
このファイルの"original"タブのB4に振動データを貼り付けると上手く行ったり行かなかったりする。貼り付けただけではセルが区切られないという場合は、"データ→区切り位置→カンマやタブなどの区切り文字によってフィールドごとに区切られたデータ→スペースにチェック"というような手順で上手くいくかもしれない。
なお、"分子の対象種を示す記号"というのがあるのだけど、今回使ったアウトプットファイルには全て"A"としかなかったので、全く意味を理解しないまま閑却しており、意味不明の記号として扱ってしまった。ここに"A"以外の記号が入るとバグるので適宜修正すると良いと思う。僕も今後同様の作業をすることがあったら修正したい。
この表のデータのうちIRチャートを書くのに必要な波数と強度だけを抜き出してシートの右側に纏めている。
"original"のタブで入力したデータは隣の"scaled"タブで呼び出して波数にスケーリングファクターを掛けている。計算によって得られる基準振動などは実測値よりも過大評価されているため、スケーリングファクターと呼ばれる数値を掛けた値がスペクトルの帰属に用いられる[2]。
なお、基底関数ごとのスケーリングファクターは以下の通り[2][3][4]。
| 基底関数 | スケーリングファクター |
| AM1 | 0.9532 |
| PM3 | 0.9761 |
| HF/3-21g | 0.9085 |
| HF/3-21g(d) | 0.8953 |
| HF/6-31g(d) | 0.8929 |
| HF/6-311g(d,p) | 0.9054 |
| MP2(full)/6-31g(d) | 0.9472 |
| MP2(fc)/6-31g(d) | 0.9434 |
| MP2(fc)/6-31g(d,p) | 0.9496 |
| SVWN/6-31g(d) | 0.9833 |
| QCISD(fc)/6-31g(d) | 0.9538 |
| BLYP/6-31g(d) | 0.9945 |
| BP86/6-31g(d) | 0.9914 |
| B3LYP/6-31g(d) | 0.9614 |
| B3P86/6-31g(d) | 0.9558 |
| B3PW91/6-31g(d) | 0.9573 |
スケーリングしたデータを"IR"タブでそれらしいIRスペクトルのチャートとして描く。この部分はIRスペクトルを描きたいで語った通り。
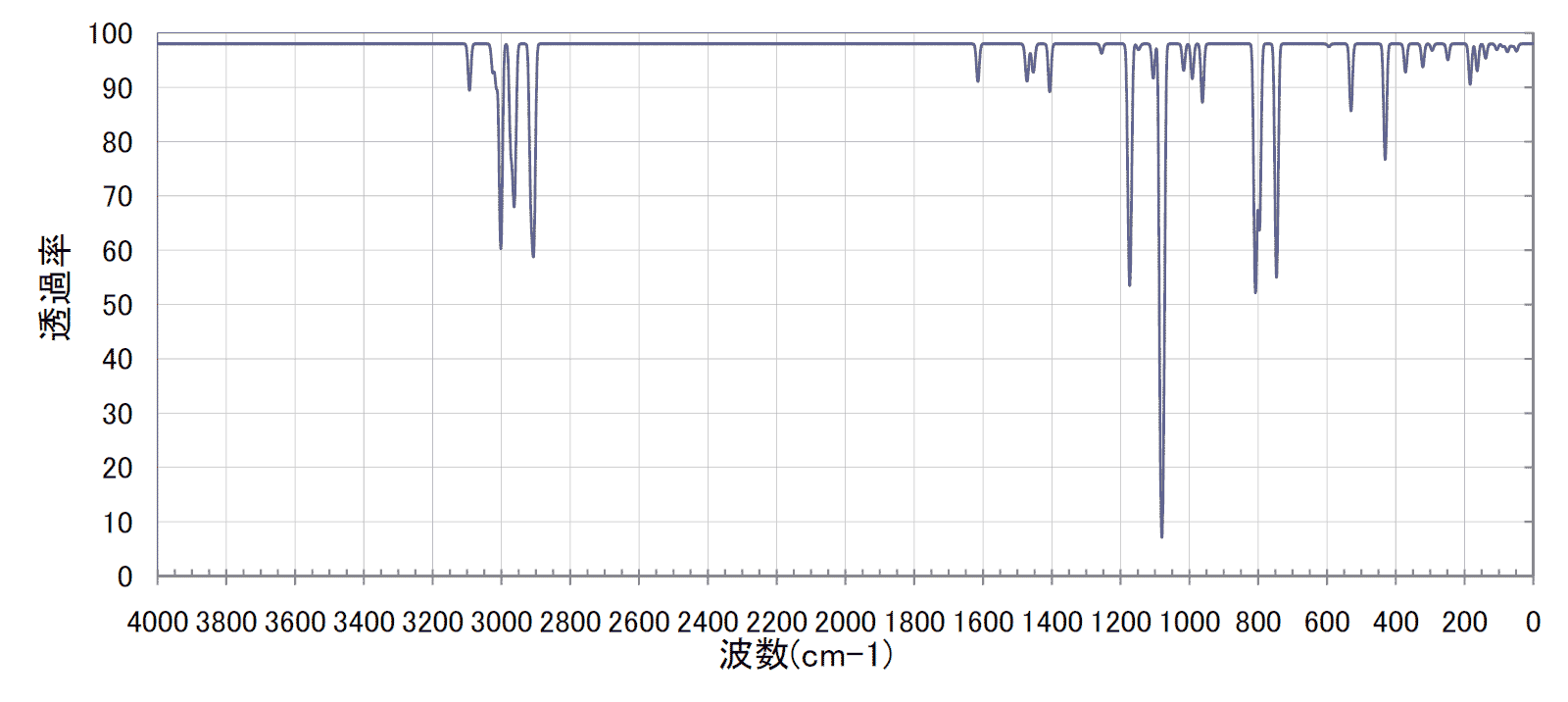
一応、原子数は27個まで対応してるけど、それ以上にしたかったらコピペとかでセルを増やしたらいくらでも増やせるようになっている。
さて、こうして計算結果を出力することができたのだが、実測値と重ね書きして比較したい。
とにかく実測値を準備しなければならない。IRスペクトルを測定すればよいのだけど、折角なので最後まで体を動かさずにやりたい。
物質のデータなんてのは大抵はデータベースにアーカイブされているもんで、探すと結構出てくる。Bio-Rad(魚拓)みたいな有料サービスもあるがブログのネタなんぞのために契約するものでもないので、今回は産総研のSDBSを使う。
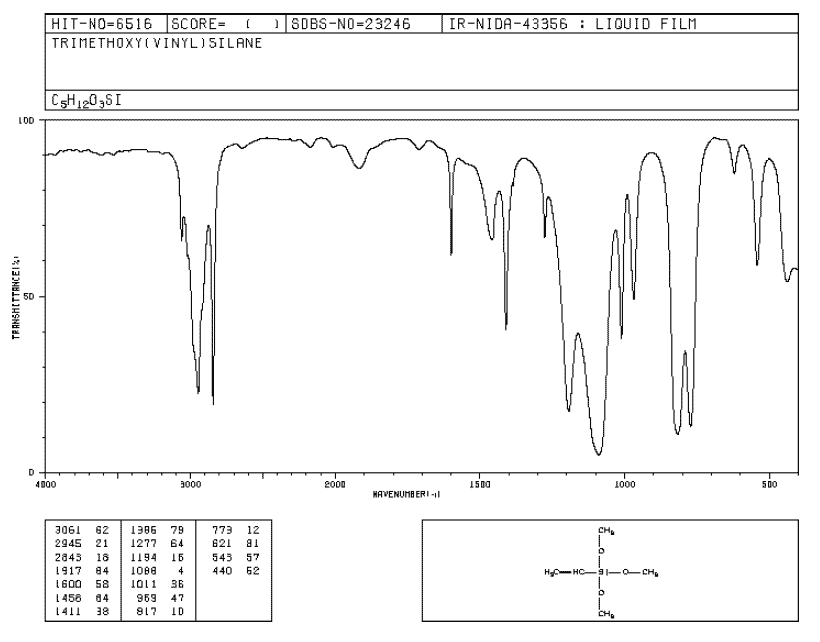
このチャートを数値化してエクセルに取り込みたい。気をつけなければならないのは、この画像横軸が2000cm-1の前と後でスケールが異なるのである。だから、数値の取り込みの際に2000より右と左で2回に分けないといけない。
以前、DigitalCurveTracerというソフトを紹介したけど、今回はWebPlotDigitizerを使う。というのはDigitalCurveTracerでは上手く数値を取り込めなかったためである。
WebPlotDigitizerの使い方はこちら(魚拓)を参考にした。
結局、得られたデータをグラフにすると下のようになった。

上手く取れていない。シャープなピークのあるグラフを取り込むのは苦手らしい。この調子だとIRだけじゃなくNMR、MS、XRDなんかに対しても使えないのが予想できる。
有用そうな類似プログラムを探すのも面倒、というか、存在するかどうかわからないものを探すのも気が進まないので、今回は画像処理で重ね書きすることにした。
上で書いたように、SDBSのIRチャートは2000cm-2を境にスケールが変化しているので、2000より上と下で2回に分けて画像を読み込んで画像を重ね、スケールを合わせて半透明化した。
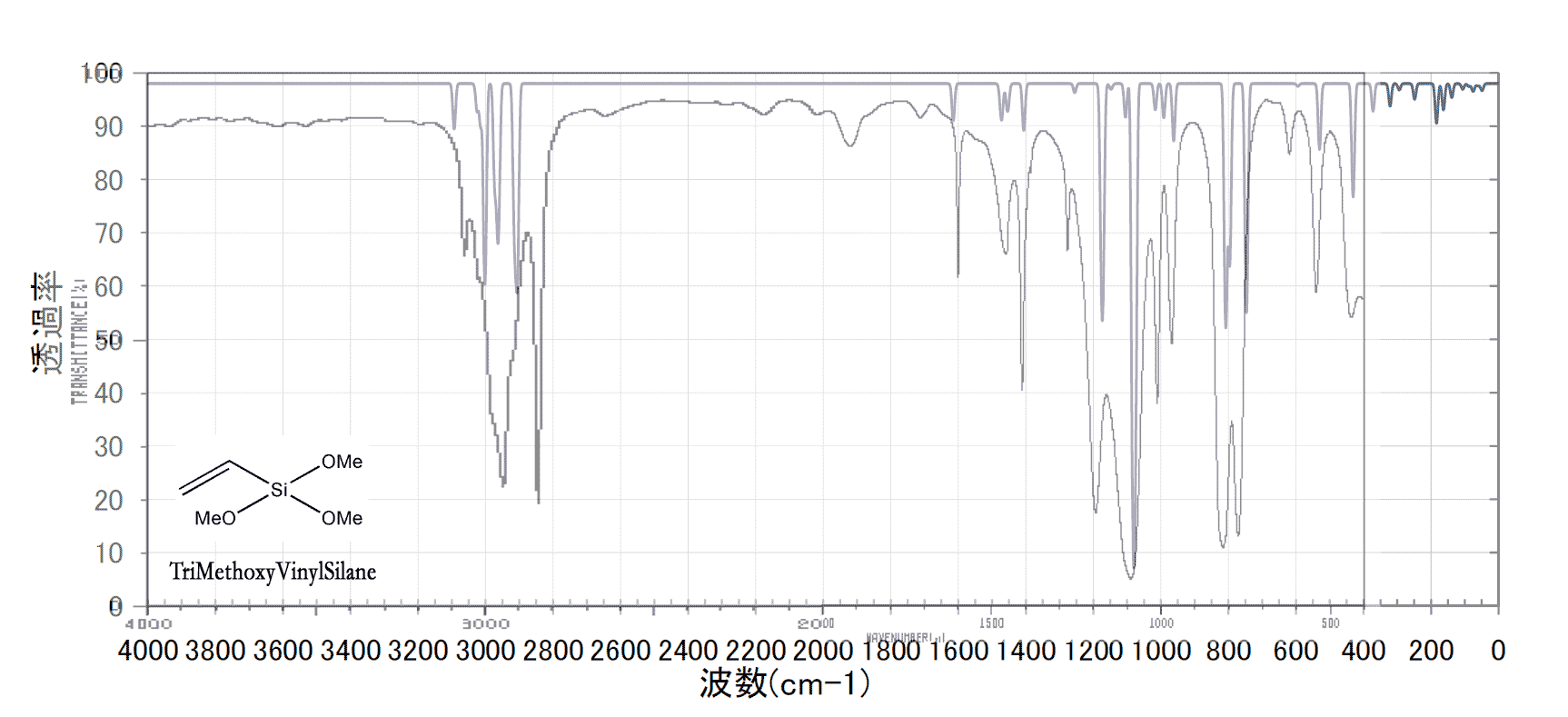
ちょっと色使いが悪いのだけど、シャープなピークが多いのが計算値で、解像度の悪いのが実測値である。結構よく一致している。
計算値のピークがどの振動を表しているかは基準振動を示すベクトルを読み込めばなんとなく見えてくる。
例えば、最も強い吸収である26番目の1081cm-1の565.127km/molは次のようになっている。
| Frequencies | -- | 1124.7548 | ||
| Red. | masses | |||
| Frc | consts | |||
| IR | Inten | 565.127 | ||
| Atom | AN | X | Y | Z |
| 1 | 6 | |||
| 2 | 6 | |||
| 3 | 1 | |||
| 4 | 1 | |||
| 5 | 1 | |||
| 6 | 14 | |||
| 7 | 8 | 0.19 | 0.19 | |
| 8 | 8 | |||
| 9 | 8 | 0.23 | 0.2 | |
| 10 | 6 | -0.14 | -0.18 | 0.13 |
| 11 | 6 | |||
| 12 | 6 | -0.19 | -0.17 | -0.15 |
| 13 | 1 | -0.23 | -0.14 | |
| 14 | 1 | 0.15 | 0.22 | -0.17 |
| 15 | 1 | -0.18 | -0.26 | |
| 16 | 1 | |||
| 17 | 1 | |||
| 18 | 1 | |||
| 19 | 1 | -0.31 | -0.17 | |
| 20 | 1 | 0.22 | 0.19 | 0.19 |
| 21 | 1 | -0.21 | -0.25 |
これを読むのはなかなか根気がいるのだけど、丁寧に読むとO7とO9のメトキシ基の振動というのがわかる。ベクトルを矢印で表すと下の絵のようになる。実際は3次元なので、このように絵を描くだけでなく、頭の中で3次元で原子の振動を描かないとちゃんとした振動の再現にはならない。
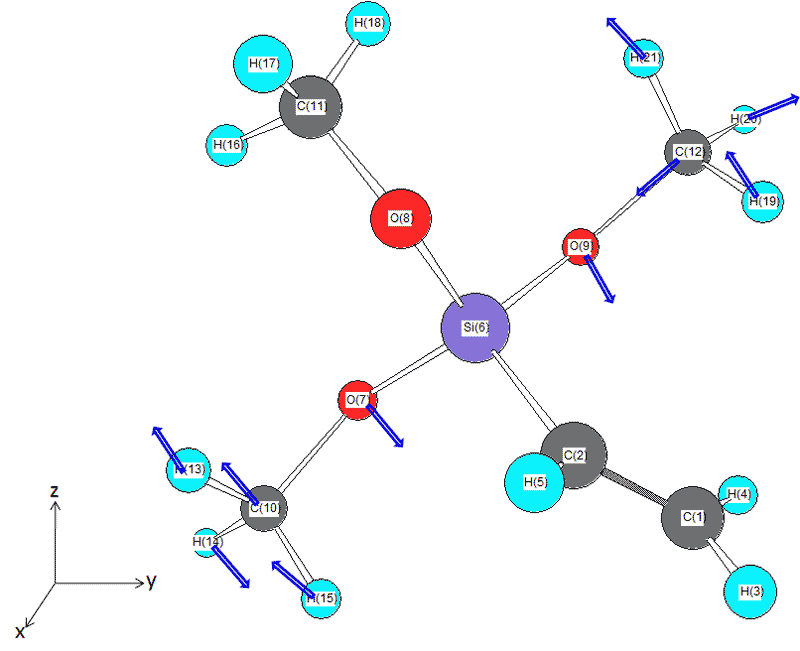
これが実測値にある1088cm-1の吸収の主な正体である。
この手続きを57回行うことで全てのピークを帰属することができる。もちろん通常は400cm-1以下は測定しないので15個くらいは切り捨てることができる。
教科書に「難しい」って書かれると、吸収帯一覧を眺めながら、あーでもないこーでもないと頭を悩ますのをイメージして、やってできないこともないけど現実的には不可能みたいなニュアンスを感じるけど、この通り振動を一つずつ解析すればそれほど無茶なことでもないのである。
参考文献
[1]マクマリー, 有機化学(上)第4版p439, 東京化学同人(1998)
[2]堀憲次, 山本豪紀, Gaussianプログラムで学ぶ情報科学・計算科学実験p57, 丸善(2006)
[3]古賀伸明, 第5版 実験化学講座12 計算化学p90, 丸善(2004)
[4]James B. Foresman, AEleen Frisch, 電子構造論による科学の探求 第2版, ガウシアン社(1998)
関連エントリー
20190621 グラフの画像から数値を読み取る
20171107 IRスペクトルを描きたい